Probing Surfaces In Realistic Conditions: AFM In Controlled Atmospheric Environments
- 10 Nov 2025
- Volume 29
- NANOscientific Magazine, 2025
Dr. Patrik Schmutz, Empa – Swiss Federal Laboratories for Materials Science and Technology Adapted from Presentation, Edited by NanoScientific
Introduction: AFM at the Interface of Reality and Control
At the 2024 NanoScientific Forum Europe, Dr. Patrik Schmutz brought the audience into the intricate world of surfaces as they exist in real environments—not just in the clean confines of a laboratory. His research at Empa focuses on the electrochemical processes and surface reactivity of metals and alloys under conditions that mimic industrial and environmental realities.
Atomic Force Microscopy (AFM) is already renowned for its ability to probe nanoscale topography and material properties. But for corrosion science, surface functionalization, and interface chemistry, the challenge lies in reproducing the environment in which a material truly operates—where humidity, temperature, and contaminants interact in complex ways.
Dr. Schmutz’s group has taken AFM into this space by modifying instruments to operate under precisely controlled atmospheric conditions. These environmental AFM experiments allow the direct study of solid–liquid interfaces—even when the “liquid” exists as a monolayer of adsorbed water or a thin electrolyte film—and the electrochemical processes that arise.
The result is a unique capability: nanoscale electrochemical reactivity measured in a realistic, tunable environment, directly linked to material performance in service.
From Lab to Field: Why Environment Matters
In industrial and natural environments, materials rarely encounter perfectly dry or perfectly wet conditions. Instead, they operate in fluctuating humidity, exposed to aggressive ions, pollutants, and temperature cycles. Even a single monolayer of adsorbed water can generate a solid–liquid interface and profoundly alter electrochemical potentials, adhesion, wettability, and surface energy.
“AFM is not just a topography tool,” Dr. Schmutz reminded the audience. “It can track electrical, magnetic, mechanical, and wetting properties—but these are strongly environment-dependent. To study them realistically, we need environmental control.”
His group works extensively with electrochemical processes at metallic surfaces, including oxidation, corrosion, and functionalization of reactive as well as medical-grade alloys. Over the years, they have adapted commercial AFMs to make use of small, sealable environmental chambers capable of regulating humidity, temperature, inert gas atmosphere, and even dosing with reactive gases or ions.
These modifications make it possible to bridge the gap between macroscopic electrochemical measurements (such as impedance spectroscopy) in bulk electrolytes and nanoscale phenomena at the solid–liquid interface.
Measuring Surface Potential in a Thin Water Layer
One focal point of Dr. Schmutz’s presentation was surface potential mapping using Scanning Kelvin Probe Force Microscopy (SKPFM)—often referred to simply as KPFM in the AFM community—under controlled atmospheric conditions.
At the heart of this approach is the recognition that the first few molecular layers of water on a metal surface define much of its electrochemical behavior.
(Fig. 1A). Even at moderate humidity, a stable water monolayer forms, with a steep potential gradient across the Helmholtz plane.
In the presence of liquid layers, KPFM can detect these potential differences best by lifting a conductive AFM tip above the surface—avoiding direct disturbance of the delicate water layer—and applying a bias to nullify the electrostatic forces. When such a nanometer-thick solid–liquid interface is formed by a controlled exposure to humidity or water, it can be demonstrated that the potential measured (referred to as the Volta potential) on pure metals correlates linearly with the Open Circuit Potentials (OCP) obtained versus a well-defined Hg/HgCl Calomel electrode (SCE) in bulk water. The measured potential contains, in both cases (KPFM and bulk), a significant charge contribution from the nanometer-thick passive oxides (Fig. 1A).
For thicker oxides, space charge effects then become a significant contribution to the solid–liquid interface potential gradients. Fig. 1B presents the example of two trends in thick (see cross-section after 100 V anodizing) anodically grown amorphous barrier oxides displaying various space-charge contributions. KPFM allowed investigation of charge buildup as a function of oxide defect densities.
In the presented example, KPFM characterization identified a remarkable difference in the potential evolution for oxides grown on 99.99% and 99.5% pure aluminum. The very high potentials measured for the 99.5% Al are attributed to denser oxide with less defect migration ability during the high-field growth anodizing process compared to the lower-density oxides formed on 99.99% Al [3]. This “density” difference has been cross-validated by other characterization methods [3].
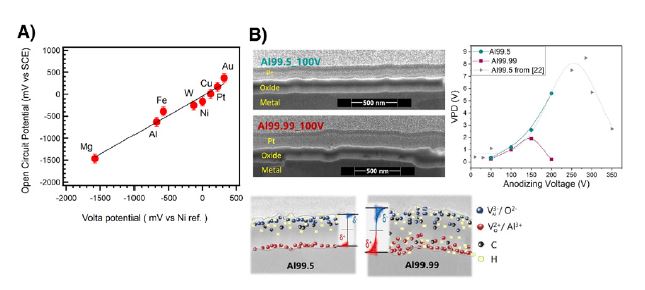
This detailed analysis of the different contributions in the solid-liquid interface potential gradients can obviously be best used with the environmental AFM /KPFM combination on micro and nanosized heterogeneous materials or components. For multiphase alloys, such as aluminum alloys with intermetallic particles (see SEM characterization in Fig. 2A), this potential mapping reveals nanoscale galvanic coupling: magnesium-rich particles appear at low potential (anodic sites), while copper- or iron-rich intermetallics appear at high potential (cathodic sites). Being able to evidence these different "surface reactivities" is essential for predicting corrosion.

Environmental Transitions: Dry, Humid, and Wet Regimes
The previously presented examples obviously already require well defined environmental control. Going one step further in the understanding of surface reactivity and degradation mechanism can be achieved by systematically varying humidity. Dr. Schmutz’s group has shown how the electrochemical behavior of multiphase materials changes across three environmental regimes:
• Dry regime (below ~20% RH): Surfaces are covered only with oxygen and oxide layers. KPFM shows strong potential contrast between phases, but the absolute potentials carry little electrochemical meaning.
• Humid regime (20–70% RH): A monolayer of water forms, enabling a solid-liquid interface with a Helmholtz-like charge gradient to build up. Potential values drop toward relevant electrochemical equilibrium, but galvanic interactions between phases are still not active evidenced by the fact that potential contrasts are still present between phases.
• Wet regime (>70% RH): A thicker continuous electrolyte layer forms. Potentials across the surface homogenize as galvanic coupling equilibrates the system. In many materials, this transition from humid to wet conditions marks the onset of aggressive corrosion. This humidity threshold is, for example, very critical for electronic micro and nanodevices.
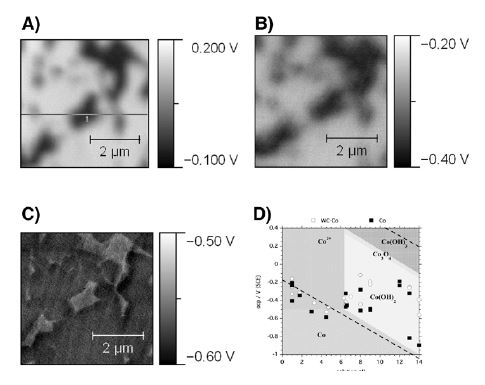
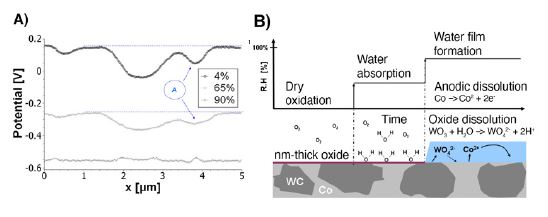
To highlight this analysis, one striking example is represented by tungsten carbide–cobalt hardmetals composites (Fig. 3) used, for example, in cutting tools and drill bits. While tungsten carbide is chemically stable in neutral environments, cobalt binder phases are highly reactive already in neutral water and slightly acidic conditions (Fig. 3D). In dry conditions (Fig. 3A), and as mentioned previously, oxidation takes places, but no solid-liquid interfaces are formed and the high measured potentials for Co (dark areas) and WC (bright areas) are not representative of electrochemical conditions. At intermediate humidity (Fig. 3B), cobalt becomes electrochemically active, the KPFM measurements indicates around -0.4 V close to the thermodynamic redox potential for the Co2+-Co equilibria as shown on the Pourbaix diagram (Fig. 3D). In wet conditions (Fig. 3C: 90% RH) Co is dissolving rapidly driven by galvanic coupling as indicated by the uniform and low potential (Co Redox potential) measured by KPFM and aggravated by the larger surface area of the carbide grains—degrading the material’s mechanical integrity.
This evolution and the related surface reaction mechanisms are then summarized in Fig. 4. The initially very large potential difference measured upon dry oxidation (4% RH), experiences first a shift toward more negative "electrochemically" relevant potentials in humid environments (65%) before a complete homogenizing and galvanic coupling is taking place in wet environments (90% RH). Dissolved ions contribute to a strengthening of this coupling effect by increasing the ionic conductivity and the nm-thick water layer.
Coupling AFM with Local Electrochemical Probes
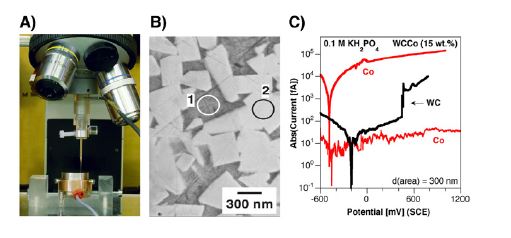
While KPFM reveals potential distributions, it does not provide direct kinetic data. For this, Dr. Schmutz’s group employs complementary techniques such as Electrochemical nanocapillary methods—a point-by-point method in which a nanocapillary containing electrolyte is positioned on the surface (Fig. 5A).
By polarizing individual phases separately, this capillary-based method can measure local anodic and cathodic currents, revealing why certain phases corrode faster than others. Following on the experiments presented in Fig. 3, the electrochemical polarization curves performed with a 300 nm diameter capillary on the Co and WC phases (Fig. 5B) confirmed that, cobalt in hardmetals exhibits high anodic dissolution currents, while tungsten carbide shows low cathodic current density.
Two measurements for different Co areas are displayed for the slightly acidic electrolyte (0.1M KH2PO4) used (Fig. 5C): one showing very high activation currents and the other one with very low passive currents. These two very different behaviors are however expected given the fact that cobalt lies in this electrolyte at its stability boundary (see Pourbaix diagram, Fig. 3D).
The extremely important information obtained in Fig. 5C by this additional characterization is:
• First in the electrochemical potential measurements. The values of around -0.2V for WC and -0.4V for Co are totally in line with the KPFM characterization in humid environment.
• The reaction kinetics indicate that the anodic dissolution of Co is much faster compared to the cathodic reduction on WC—confirming the non-polarizable, sacrificial role of cobalt in galvanic coupling justifying that the whole surface is polarized to its low potential.
This local kinetic information obtained by the nanocapillary technique is essential for understanding how isolated defects or inclusions can dictate the corrosion behavior of an entire component, but the KPFM analysis was in fact sufficient to document the surface reactivity mechanisms and Fig. 3. Relative humidity (RH) the non-polarizable nature of the Co areas!
Industrial Case Study: Blistering of Aluminum-Tin-Coated Bearings
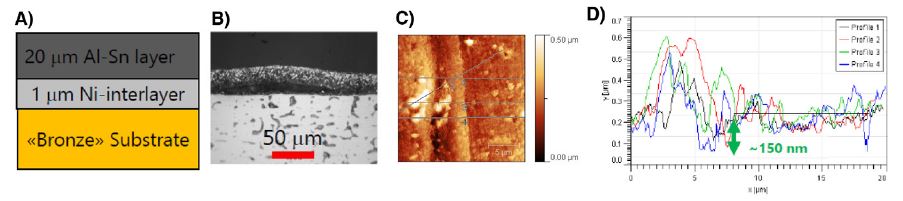
The power of environmental AFM becomes clear in industrial case studies. Dr. Schmutz presented the example of large bearings with an Al-Sn top layer over a nickel interlayer and bronze substrate (Fig. 6A). The tin layer acts as a solid lubricant, reducing friction in service. However, in certain polluted climates, bearings developed surface blisters after only a year in storage. Cross-sectional analysis revealed localized delamination, typically around 100 μm, and corrosion beneath the coating (Fig. 6B).
Environmental AFM measurements of fresh coating cross-sections showed that under high humidity (90% RH), tin rapidly oxidizes and migrates away from the interface. When sulfur-containing pollutants were additionally introduced, the oxidation products became voluminous, causing mechanical disruption of the coating. KPFM maps showed localized high-potential regions adjacent to reactive dissolving zones—conditions ideal for galvanic corrosion.
The degradation rate measured in the laboratory under environmental control—about 150 nm trenching at the interface (Fig. 6C, 6D) after 18 hours of aggressive environment exposure—might not seem large, but it closely matched the field-observed blister growth rate of 70 μm/year. The conclusion was clear: failure could only be reproduced in this environment and required the combined effect of high humidity and sulfur pollution, conditions common in industrial polluted environments.
Conclusion: AFM as a Window into Real-World Interfaces
Through carefully controlled atmospheric experiments, Dr. Schmutz’s group has shown that AFM can go far beyond imaging. By combining topography, potential mapping, and local electrochemistry under realistic environmental conditions, AFM becomes a predictive tool for material performance.
Key takeaways from his work:
• Realistic surface reactivity and electrochemical behavior emerge only when a solid–liquid interface exists—even as a thin adsorbed film.
• Surface potential mapping correlates closely with macroscopic corrosion tendencies if environmental conditions are well defined during the AFM characterization.
• Transitions between dry, humid, and wet regimes are critical thresholds for corrosion initiation of heterogeneous materials and components.
• Industrial failures often involve environmental synergies, such as humidity plus pollutants, which can be reproduced and quantified in controlled closed chambers in the laboratory.
As Dr. Schmutz summarized for the NSFE audience, “An AFM with environmental control lets us look at surfaces the way they live in the real world.” This approach is proving indispensable for industries where durability, corrosion resistance, and functional coatings determine long-term performance.

Dr. Patrik Schmutz is a leading researcher and Group Leader in the Joining Technologies & Corrosion laboratory at Empa (empa.ch)—the Swiss Federal Laboratories for Materials Science and Technology—located in Dübendorf, Switzerland. He also holds a lecturer position in Materials Science at ETH Zürich (ch.linkedin.com). He obtained his Ph.D. degree from EPF Lausanne, Switzerland, followed by post-doctoral research stays at Ohio State University and ETH Zürich.
Dr. Schmutz leads research on electrochemical oxidation/passivation and medical implant surfaces, focusing on the reactivity and stability of functionalized surfaces in real-world environments—from humid, polluted atmospheres to complex biological media. His group employs a broad suite of techniques, including environmental AFM (notably SKPFM), local electrochemical methods, impedance spectroscopy, electrochemical quartz microgravimetry, HAXPES/XPS, and spectroscopic imaging, to interrogate processes like passivation, galvanic coupling, and corrosion kinetics in advanced materials and biomedical implants.
His work spans both fundamental understanding and applied solutions in industries ranging from microelectronics and aerospace to civil engineering and healthcare. For more information, contact patrik.schmutz@empa.ch or visit Empa – Joining Technologies and Corrosion – Surface Electrochemistry.
References
-
P. Schmutz, G. S. Frankel, “Characterization of AA2024-T3 by Scanning Kelvin Probe Force Microscopy,” Journal of the Electrochemical Society, 145(7), 2285–2295 (1998).
-
V. Guillaumin, P. Schmutz, G. S. Frankel, “Characterization of Corrosion Interface by the Scanning Kelvin Probe Force Microscopy Technique,” Journal of the Electrochemical Society, 148(5), B163–B173 (2001).
-
M. González-Castaño, M. Döbeli, V. Araullo-Peters, L. P. H. Jeurgens, P. Schmutz, C. Cancellieri, “Substrate Purity Effect on the Defect Formation and Properties of Amorphous Anodic Barrier Al₂O₃,” Journal of the Electrochemical Society, 165(7), C422–C431 (2018).
-
F. Evangelisti, M. Stiefel, O. Guseva, R. Partovi Nia, R. Hauert, E. Hack, L. P. H. Jeurgens, P. Schmutz, C. Cancellieri, “Electronic and Structural Characterization of Barrier-Type Amorphous Aluminium Oxide,” Electrochimica Acta, 224, 503–516 (2017).
-
S. Hochstrasser (Kurz), C. Latkoczy, D. Günther, S. Virtanen, P. J. Uggowitzer, P. Schmutz, “ICP-MS, SKPFM, XPS and Microcapillary Investigation of the Local Corrosion Mechanisms of WC-Co Hardmetal,” Journal of the Electrochemical Society, 155(8), C415–C426 (2008).
Category